A very long time ago I asked whether Neandertals’ brains grew like ours do today, a question raised by conflicting results coming from two research teams. Both teams reconstructed the brain endocasts of modern humans and fossil Neandertals, and compared how endocast shapes changed during growth and development. As I mused in that post, the different results seem to result largely from differences in how a critical fossil specimen (the Neandertal newborn from Mezmaiskaya, Russia) was reconstructed.
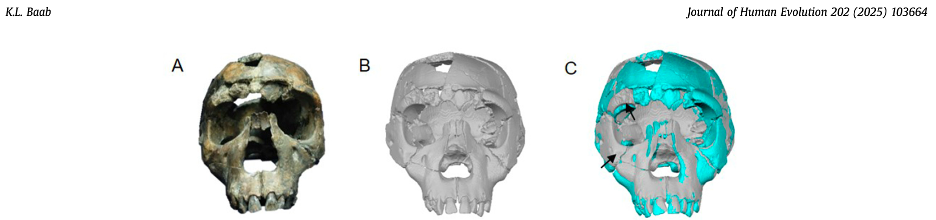
This is a perennial problem for paleoanthropology. Our knowledge of the human past hinges on a few thousands of individuals whose bones and teeth managed to survive and be discovered after several thousands or millions of years. Most of these precious remains are fragmentary and cannot speak for themselves. So, researchers must rely on their own anatomical expertise and a bit of artistic license to reconstruct what many key fossils would have looked like in their original condition.
Over thirty years ago Christophe Zollikofer and colleagues (1995: 283) reported that, “Fossil specimens can be restored, measured and replicated without physical contact using … computer assisted reconstruction.” The development of these “virtual anthropology” methods has made fossil reconstruction much more accessible. Most importantly, virtual methods allow researchers to generate multiple, reasonably realistic reconstructions of the same fossil. As Philipp Gunz and colleagues (2009: 61) noted, “While there typically will be shape differences among equally plausible reconstructions, these different estimates might still support a single conclusion. But they need not do so, and all assumptions must be strenuously challenged if one or more reconstructions, or a statistical analysis based on them, are to be treated as arguments for a scientific claim.”
As these paleo pioneers have also acknowledged, making data publicly available will also help assess the extent to which specific reconstructions might affect subsequent interpretations. Both of these research groups have published 3D landmark datasets with some overlapping specimens, allowing us to address this central question. Simon Neubauer and colleagues (2018) published the landmark data used in their reconstruction and analysis of a juvenile Homo erectus cranium (here). A team led by Marcia Ponce de León (2021) and Christophe Zollikofer (2022) have posted comparable data from their endocast reconstructions of Homo erectus from Dmanisi, Georgia (here) and early Homo sapiens from Herto, Ethiopia (here). These great datasets bear on the evolution of brain size and shape—let’s dig in.
Both groups—Neubauer et al. and Ponce de León et al. + Zollikofer et al. (hereafter “PZ”)—include recent modern humans from different skeletal collections and the same nine fossil Homo specimens: KNM-ER 1813 (H. habilis), KNM-ER 1470 (H. rudolfensis), and seven other fossils from Kenya and Indonesia typically attributed to Homo erectus. Most of the fossils required varying extents of reconstruction, from the alignment of separate cranial fragments to the mathematical estimation of endocranial surfaces that aren’t preserved. The two teams measured endocast shape using comparable but slightly different sets of 3D landmark coordinates, so we can’t combine the datasets but we can run the same set of analyses on each sample separately and then compare the results.
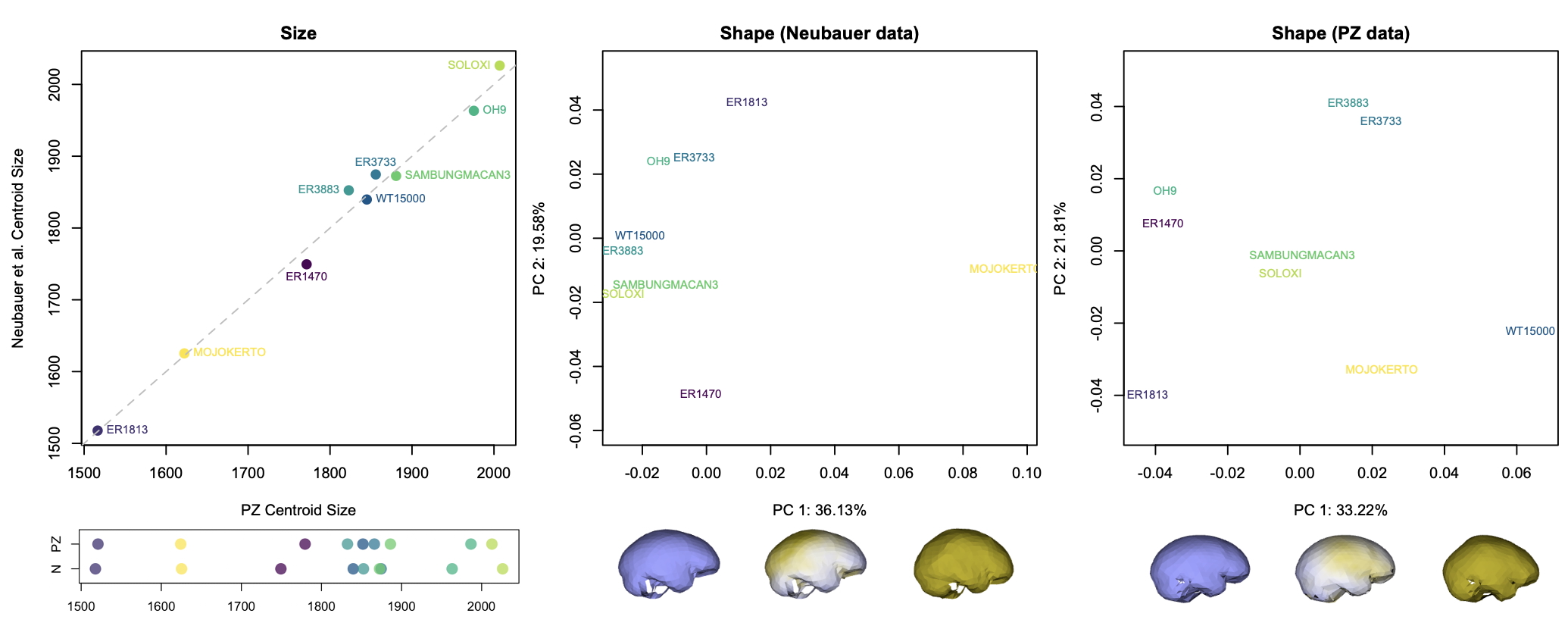
The graphs above show how the nine fossils vary within and between datasets. The 3D landmarks used to measure endocast size and shape return similar overall sizes for each specimen (left graphs). There are differences in the relative positions of a few specimens (ER 3883 vs. WT 15000 and ER 3733 vs. Sambungmacan 3), but these discrepancies are small probably mostly within the range of uncertainty for individual fossil reconstructions.
The effects of different reconstructions on endocranial shape, on the other hand, are a bit more profound. In each dataset, the main dimension of variation (PC1, the horizontal axis in the center and right graphs) captures similar patterns of shape variability. In both samples, fossils with a longer and lower endocast fall on the left side of the graph, while rounder endocasts fall on the right side of the graph. But where individual specimens plot in the graphs (i.e., their overall endocast shape) differs notably between datasets. For example, the “Mojokerto” infant Homo erectus has the roundest shape while WT 15000 has one of the ‘flatter’ shapes in the Neubauer sample, whereas WT 15000 is the ‘roundest’ in the PZ sample.
So, different decisions in the reconstruction process can lead to different overall patterns of shape variation within a sample. This can have important impacts on subsequent analyses. For instance, we often want to assess how similar or different fossil specimens are to one another, looking for clusters of similar shapes that might tell us something meaningful about the biology we’re hoping to capture. The two datasets, however, produce slightly different clusters:
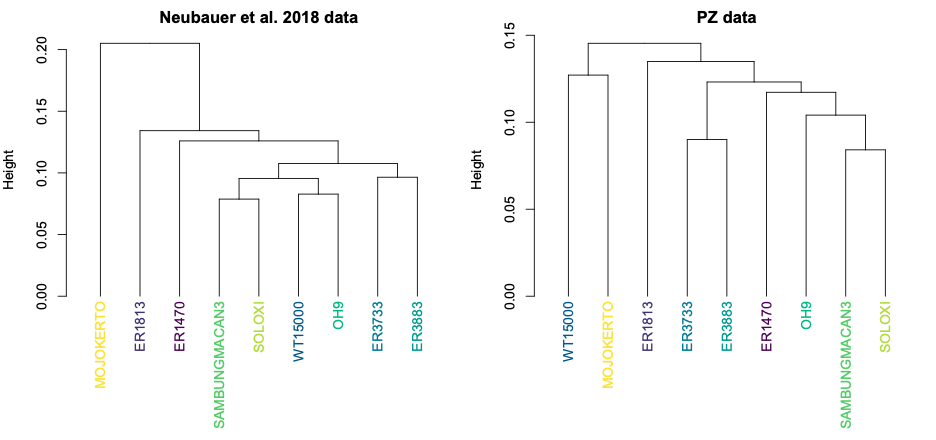
Both datasets produce clusters with early H. erectus specimens ER 3733 and ER 3883, and later Indonesian H. erectus fossils Sambungmacan 3 and Solo XI. But the similarities among other fossils differ between the two samples, in ways that could lead to different biological interpretations. One might interpret the Neubauer clustering to mean that the Mojokerto infant differs from the rest since it hadn’t completed brain growth, while the other clusters could potentially reflect evolutionary changes both from early Homo (ER 1813 and 1470) to H. erectus and over time within H. erectus. In contrast, the PZ tree could be interpreted to mean that the adolescent WT 15000 had an ‘underdeveloped’ brain like Mojokerto, while the different clusters of ER 1813 and ER 1470 could reflect a more convoluted pattern of brain evolution from early Homo to H. erectus.
Of course, principal components and cluster analyses are statistical approaches for exploring variation within a sample, and they don’t necessarily map onto meaningful phenomena. Biological patterns could ‘override’ variation due to differences in reconstruction. For instance, endocast shape variation due to growth and development could produce marked, characteristic differences between infants and adults. Indeed, if we compare endocast shape of the infant Mojokerto to the average adult H. erectus, both datasets yield fairly similar results:
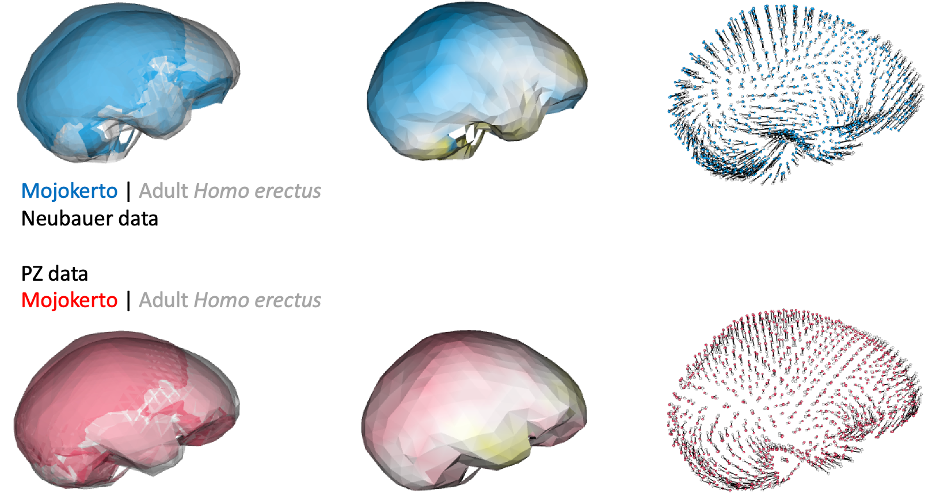
In addition, if groups/species have distinct endocast shapes, such differences could still be captured by studies using different fossil reconstructions. For instance, both studies produce similar results when comparing early Homo specimens ER 1813 and ER 1470, and comparing adult H. erectus and modern humans:
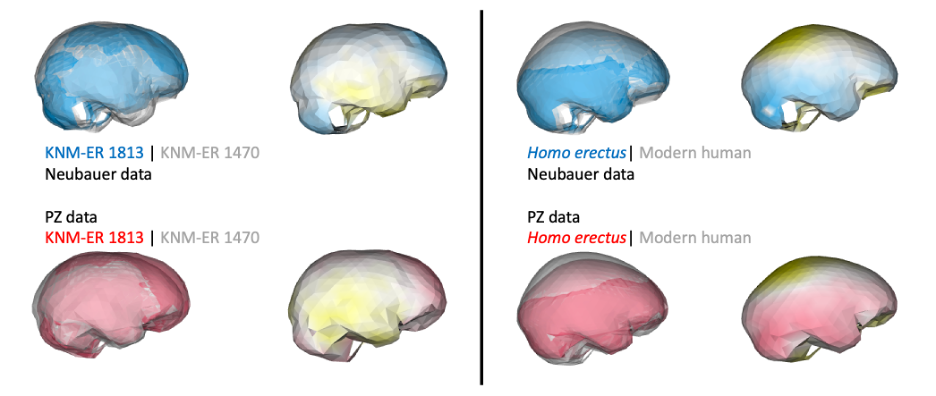
So, getting back to our original question: do different virtual reconstructions produce different results? Yes and no. Yes, there will be observable differences between studies, and these could be subtle (e.g., brain sizes estimates) or more severe (e.g., clustering patterns within a fossil sample). But as Melvin Moss reminded us, we must keep in mind the underlying biological questions when interpreting statistical patterns. Ultimately, fossil preservation is probably the greatest source of variability between different studies. Many researchers will bring similar levels of expertise and similar analytical toolkits to study fossils, but more fragmentary specimens will have greater uncertainty in how to to reconstruct them. In contrast to the different growth patterns identified in the Neandertal studies mentioned at the beginning of this post, the consistent ‘growth’ signal in H. erectus fossils may be due to the fact that the Mojokerto infant is better preserved and required less reconstruction than Neandertal neonates.
As Gunz and colleagues (2009) stressed when they laid out “principles for the virtual reconstruction of hominin crania,” these powerful virtual methods can never produce “the” single correct reconstruction of a fossil. Rather, researchers must acknowledge and remain cognizant of all the decisions and assumptions that go into their reconstructions, and attempt to produce multiple reconstructions reflecting these varied uncertainties. Making data openly available further allows other researchers to assess how conclusions were reached, and to add new fossils to existing datasets.
REFERENCES
Baab, K. L. (2025). A fresh look at an iconic human fossil: Virtual reconstruction of the KNM-WT 15000 cranium. Journal of Human Evolution, 202, 103664. https://doi.org/10.1016/j.jhevol.2025.103664
Gunz, P., Mitteroecker, P., Neubauer, S., Weber, G. W., & Bookstein, F. L. (2009). Principles for the virtual reconstruction of hominin crania. Journal of Human Evolution, 57(1), 48–62. https://doi.org/10.1016/j.jhevol.2009.04.004
Neubauer, S., Gunz, P., Leakey, L., Leakey, M., Hublin, J.-J., & Spoor, F. (2018). Reconstruction, endocranial form and taxonomic affinity of the early Homo calvaria KNM-ER 42700. Journal of Human Evolution, 121, 25–39. https://doi.org/10.1016/j.jhevol.2018.04.005
Ponce De León, M. S., Bienvenu, T., Marom, A., Engel, S., Tafforeau, P., Alatorre Warren, J. L., Lordkipanidze, D., Kurniawan, I., Murti, D. B., Suriyanto, R. A., Koesbardiati, T., & Zollikofer, C. P. E. (2021). The primitive brain of early Homo. Science, 372(6538), 165–171. https://doi.org/10.1126/science.aaz0032
Zollikofer, C. P. E., Bienvenu, T., Beyene, Y., Suwa, G., Asfaw, B., White, T. D., & Ponce De León, M. S. (2022). Endocranial ontogeny and evolution in early Homo sapiens: The evidence from Herto, Ethiopia. Proceedings of the National Academy of Sciences, 119(32), e2123553119. https://doi.org/10.1073/pnas.2123553119
Zollikofer, C. P. E., Ponce de León, M. S., Martin, R. D., & Stucki, P. (1995). Neanderthal computer skulls. Nature, 375(6529), 283–285. https://doi.org/10.1038/375283b0