A 45,000 year old human femur from Siberia provides new information about genetic mutation rates and modern human origins. As Quiaomei Fu and colleagues report in this week’s issue of Nature, this seemingly simple leg bone carries so much information, not because of its gross anatomy, but because of the ancient DNA it preserves.
The femur wasn’t discovered by paleontologists, but by an artist/historian looking for fossils around the Irtysh River. The bone came from from a site called Ust’-Ishim, only some 650 km north of the snowy capital where I work in Kazakhstan:
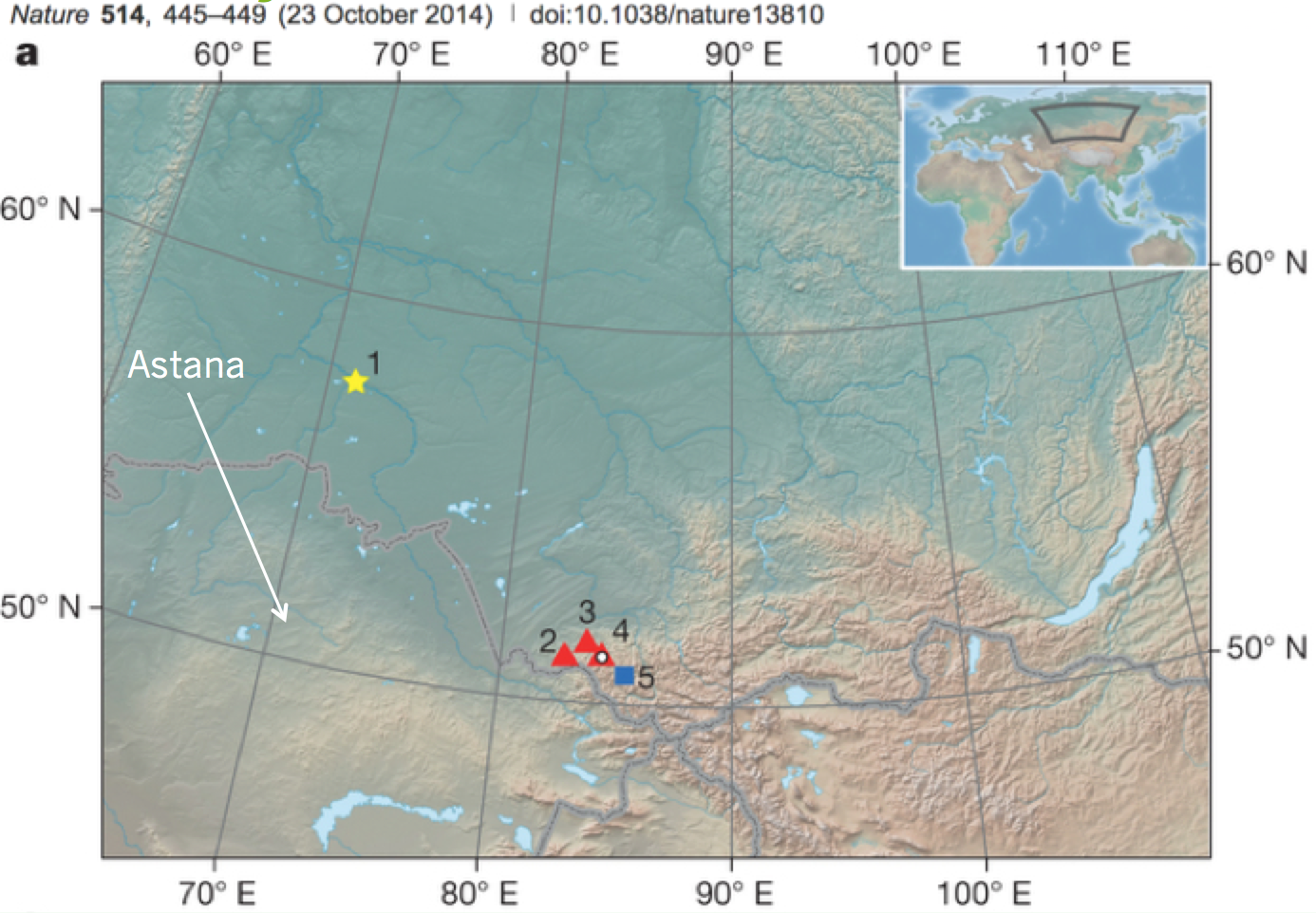
The site in question, Ust’-Ishim is marked by the yellow star. The red and blue sites to the southeast are other Upper Paleolithic sites. Okladnikov (3) and Denisova (4) have also yielded fossils preserving ancient DNA. Modified from Fu et al. figure 1.
The bone was directly radiocarbon dated to around 45,000 years ago. With a fairly precise age of the bone, Fu et al. could estimate the rate at which genetic mutations arise, by counting the number of new mutations in recent humans that aren’t shared by the Ust’-Ishim femur. This led to an estimate of around 0.43×10−9 new mutations per site per year. This is a relatively low rate compared to estimates based on geologically older fossils, but consistent with more recent estimates that directly compare parents and offspring.
The Ust’-Ishim individual had levels of Neandertal ancestry comparable to living Eurasians (~2.3% of the genome), but there is no evidence of any Denisovan ancestry. Because this individual lived closer to the date of modern-Neandertal admixture, the Neandertal segments of its genome are longer than in modern people (recombination over generations breaks these regions apart into shorter segments). Knowing about recombination rates, Fu et al. could infer that admixture between Neandertal and modern human populations occurred between 50-60,000 years ago.
This eFfing Friday fossil provides more tantalizing evidence for DNA-bearing human fossils just across the Kazakhstan border. With Ust’-Ishim to the north, Denisova and Okladnikov caves to the east, and Teshik Tash to the south, my colleagues and I are very keen to find similar sites here on the KZ side.
Reference: Fu et al. 2014. Genome sequence of a 45,000-year-old modern human from Siberia. Nature 514: 445–449. doi:10.1038/nature13810.


