For last week’s AAPA conference, my friend and colleague David Pappano organized a workshop teaching about the many uses of the R programming language for biological anthropology (I’m listed as co-organizer, but really David did everything). After introducing the basics, we broke into small groups focusing on specific aspects of using R. I devised some lessons for basic statistics, writing functions, and resampling. Since each of the lessons could have easily taken up an hour and most people didn’t get to go through the activities fully, I’m posting up the R codes here for people to mess around with.
The basic stats lesson utilized Francis Galton’s height data for hundreds of families, courtesy of Dr. Ryan Raaum. To load in these data you just need to type into R: galton = read.csv(url(“http://bit.ly/galtondata“)). The code simply shows how to do basic statistics that are built into R, such as t-test and linear regression.

Some summary stats for the Galton data. The code is in blue and the output in black.
Here is the Basic Stats code, download and paste it into an R file, then buckle up!
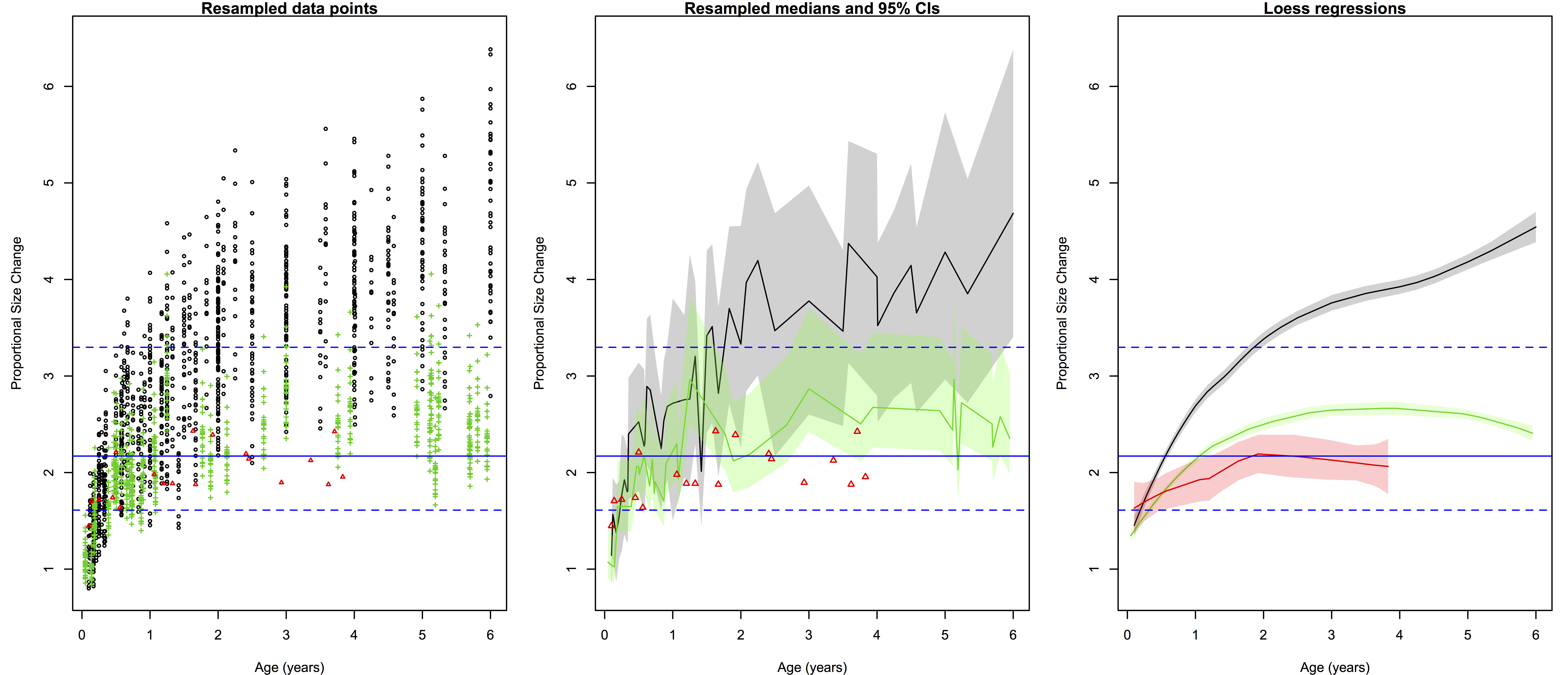
The lesson on functions and resampling was based on limb length data for apes, fossil hominins and modern humans (from Dr. Herman Pontzer). The csv file with the data can be downloaded from David’s website. R has lots of great built-in functions (see basic stats, above), and even if you’re looking to do something more than the basics, chances are you can find what you’re looking for in one of the myriad packages that researchers have developed and published over the years. But sometimes it’s necessary to write a function on your own, and with fossil samples you may find yourself needing to do resampling with a specific function or test statistic.
For example, you can ask whether a small sample of “anatomically modern” fossil humans (n=12) truly differs in femur length from a small sample of Neandertals (n=9). Traditional statistics require certain assumptions about the size and distribution of the data, which fossils fail to meet. Another way to ask the question is, “If the two groups come from the same distribution (e.g. population), would random samples of sizes n=12 and n=9 have so great an average difference as we see between the fossil samples?” A permutation test, shuffling the group membership of the fossils and then calculating the difference between the new “group” means, allows you to quickly and easily ask this question:

R code for a simple permutation test. The built-in function “sample()” is your best friend.
Although simply viewing the data suggests the two groups are different (boxplot on the left, below), the permutation test confirms that there is a very low probability of sampling so great a difference as is seen between the two fossil samples.

Left: Femur lengths of anatomically modern humans (AMH) and Neandertals. Right: distribution of resampled group differences. Dashed lines bracket 95% of the resampled distribution, and the red line is the observed difference between AMH and Neandertal femur lengths. Only about 1% of the resampled differences are as great as the observed fossil difference.
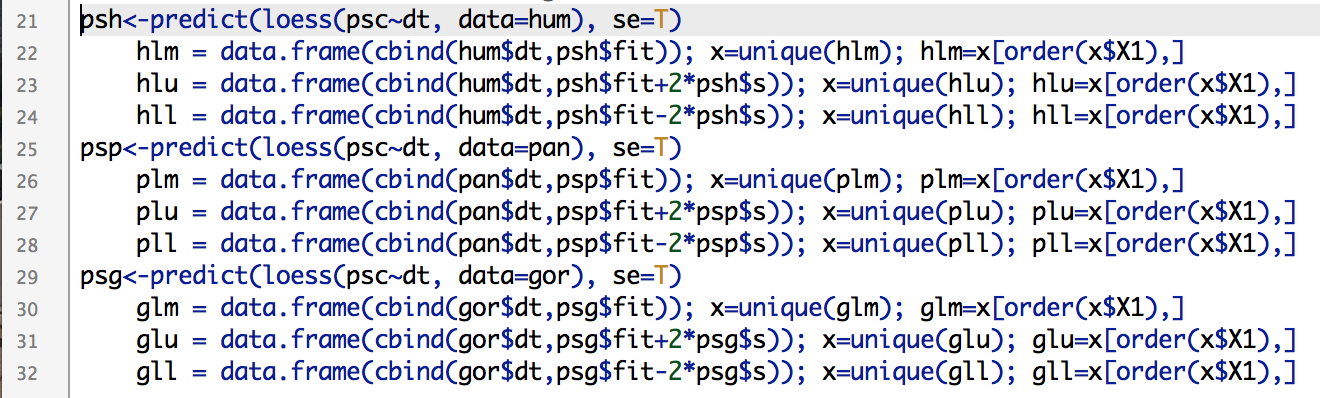
Here’s the code for the functions & resampling lesson. There are a bunch of examples of different resampling tests, way more than we possibly could’ve done in the brief time for the workshop. It’s posted here so you can wade through it yourself, it should keep you busy for a while if you’re new to R. Good luck!